If you read the papers or watch the news you’ve probably heard about the recently published JUPITER study, advertised with bold headlines such as “Cholesterol drug causes risk of heart attack to plummet” and “Cholesterol-fighting drug shows wider benefit”. If you’ve been following this blog (and perhaps even if you haven’t), you are by now aware that such claims cannot be taken at face value.
You might suspect, for example, that the study was sponsored by a drug company and authored by researchers with financial interests tied to those drug companies. You might wonder if these associations could possibly – just possibly – influence not only the results of the study, but how those results are reported. You might also find yourself questioning the objectivity of a study with the title “Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin” (JUPITER).
If you’re asking yourself these questions, you are definitely on the right track. The study was indeed sponsored by a drug company, AstraZeneca. And each author of this study received money in the form of grants, consulting fees and honoraria from pharmaceutical companies – in some cases up to twelve different companies, including AstraZeneca, the study sponsor. Take a look at this list detailing the financial interests of the study authors (now required by the New England Journal of Medicine and other prominent publications):
Dr. Ridker reports receiving grant support from AstraZeneca, Novartis, Merck, Abbott, Roche, and Sanofi-Aventis; consulting fees or lecture fees or both from AstraZeneca, Novartis, Merck, Merck–Schering-Plough, Sanofi-Aventis, Isis, Dade Behring, and Vascular Biogenics; and is listed as a coinventor on patents held by Brigham and Women’s Hospital that relate to the use of inflammatory biomarkers in cardiovascular disease, including the use of high-sensitivity C-reactive protein in the evaluation of patients’ risk of cardiovascular disease. These patents have been licensed to Dade Behring and AstraZeneca. Dr. Fonseca reports receiving research grants, lecture fees, and consulting fees from AstraZeneca, Pfizer, Schering-Plough, Sanofi-Aventis, and Merck; and Dr. Genest, lecture fees from AstraZeneca, Schering-Plough, Merck–Schering-Plough, Pfizer, Novartis, and Sanofi-Aventis and consulting fees from AstraZeneca, Merck, Merck Frosst, Schering-Plough, Pfizer, Novartis, Resverlogix, and Sanofi-Aventis. Dr. Gotto reports receiving consulting fees from Dupont, Novartis, Aegerion, Arisaph, Kowa, Merck, Merck–Schering-Plough, Pfizer, Genentech, Martek, and Reliant; serving as an expert witness; and receiving publication royalties. Dr. Kastelein reports receiving grant support from AstraZeneca, Pfizer, Roche, Novartis, Merck, Merck–Schering-Plough, Isis, Genzyme, and Sanofi-Aventis; lecture fees from AstraZeneca, GlaxoSmithKline, Pfizer, Novartis, Merck–Schering-Plough, Roche, Isis, and Boehringer Ingelheim; and consulting fees from AstraZeneca, Abbott, Pfizer, Isis, Genzyme, Roche, Novartis, Merck, Merck–Schering-Plough, and Sanofi-Aventis. Dr. Koenig reports receiving grant support from AstraZeneca, Roche, Anthera, Dade Behring and GlaxoSmithKline; lecture fees from AstraZeneca, Pfizer, Novartis, GlaxoSmithKline, DiaDexus, Roche, and Boehringer Ingelheim; and consulting fees from GlaxoSmithKline, Medlogix, Anthera, and Roche. Dr. Libby reports receiving lecture fees from Pfizer and lecture or consulting fees from AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, Merck, Pfizer, Sanofi-Aventis, VIA Pharmaceuticals, Interleukin Genetics, Kowa Research Institute, Novartis, and Merck–Schering-Plough. Dr. Lorenzatti reports receiving grant support, lecture fees, and consulting fees from AstraZeneca, Takeda, and Novartis; Dr. Nordestgaard, lecture fees from AstraZeneca, Sanofi-Aventis, Pfizer, Boehringer Ingelheim, and Merck and consulting fees from AstraZeneca and BG Medicine; Dr. Shepherd, lecture fees from AstraZeneca, Pfizer, and Merck and consulting fees from AstraZeneca, Merck, Roche, GlaxoSmithKline, Pfizer, Nicox, and Oxford Biosciences; and Dr. Glynn, grant support from AstraZeneca and Bristol-Myers Squibb. No other potential conflict of interest relevant to this article was reported.
Now that you’ve put on your “Healthy Skeptic” goggles, we can move on and more closely examine the study itself. There are several things you need to be aware of as we discuss it.
First, although the press articles claim that the study looked at statin use in healthy populations, the subjects were people who had normal cholesterol but high CRP levels. CRP, or C-Reactive Protein, is a measure of inflammation in the body. It is now widely accepted even in the mainstream medical community that inflammation is a major risk factor for heart disease. And because inflammation is a sign of an underlying disease process, these patients were not, in fact, “healthy” as claimed.
There is little doubt that statins reduce inflammation, which can help prevent atherosclerosis. It appears that the benefits of statins are mainly due to this characteristic, rather than to their cholesterol-lowering effects. So it’s no surprise that the statins reduced rates of heart disease and mortality in this population that had inflammation going into the study.
I should also mention, however, that the predictive value of CRP for heart disease is highly controversial. Though some studies show a correlation between high CRP levels and heart disease, many others do not. Many physicians feel that CRP is not a useful indicator in clinical practice.
The second thing you need to be aware of is the difference between relative and absolute risk reduction. Relative risk reduction (RRR) measures how much the risk is reduced in the experimental group compared to a control group. Absolute risk reduction (ARR) is just the absolute difference in outcome rates between the control and treatment groups.
To make this more clear, let’s consider an example. Say that 2000 people enter a study for a particular drug and 1000 of them are randomized to placebo. At the end of the study, one person in the drug group died versus two people in the placebo group. The relative risk reduction of the drug group would thus be 50% (0.002 – 0.001/0.002). That sounds really impressive! The headline for this study might read “New drug reduces chance of dying by 50%!”. While technically true, you can see how misleading this can be. Why? Because when most people read that headline, they will interpret it to mean that if they take that drug, their risk of dying will be reduced by 50%, which is not even close to being true.
The absolute risk reduction, on the other hand, is always a much more modest number. Using the same example above, the absolute risk reduction in the drug group would have been a paltry one-tenth of a percent, or 0.1% (0.002 – 0.001). That’s not a very catchy headline, is it? “New drug reduces risk of dying by one-tenth of a percent”. It just doesn’t grab you the same way. But this is actually a more realistic view of what happened in the study and what we could expect to happen in the real world.
In fact, one could just as accurately say that in this hypothetical study, a patient has a 1-in-1000 (0.1%) chance of their life being saved by the drug. Said another way, 1,000 patients would have to be treated with this drug in order to save a single life. This measurement is called the Needed Number to Treat, and is another means for interpreting the results of clinical trials.
With that in mind, let’s examine the data from the JUPITER study. The actual numbers were 198 deaths out of 8901 in the statin group and 247 deaths out of 8901 in the placebo group. The relative risk reduction for total mortality (deaths) in the drug group was 19.8% [(247/8901 - 198/8901) / (247/8901)]. That means that the risk of death for people taking Crestor was 19.8% smaller than those taking placebo.
But what happens when we look at the absolute risk reduction numbers? According to the data, 2.77% (0.02774) of people taking the placebo died after two years versus 2.24% (0.02224) of people taking Crestor. This amounts to a difference of 0.55%, or one-half of one percent.
Here’s a graphical illustration of the difference in mortality between the Crestor and placebo group:
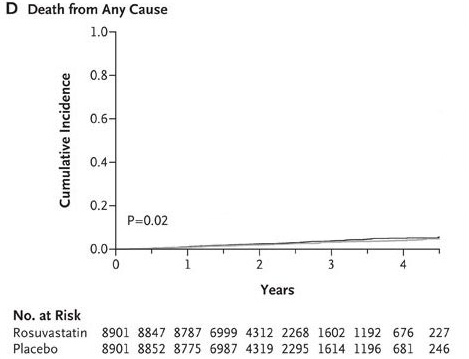
If you’re having trouble making much of a difference, I don’t blame you!
To make this even more clear, let’s use the Needed Number to Treat method of evaluating these results. According to the study data, 182 people would have to be treated with Crestor for two years in order to save a single life.
Now that may not sound like a large number to you, especially if yours was one of the lives saved. However, when evaluating the viability of any potential treatment three considerations (above and beyond the efficacy of the treatment) must be taken into account: cost, side effects, and alternatives.
Let’s look at cost first. The cost of one patient taking Crestor for one year is approximately $1,300. Therefore, to prevent 49 deaths 8,901 people would have to take Crestor for two years at a cost of $23 million dollars. That is an enormously expensive treatment by any measure.
Second, this particular study did not register significant side effects in the statin group. This is very fishy, though, since nearly every other study on statins to date has shown significant side effects and the approval of Crestor itself was delayed by the FDA due to concern about Crestor side effects.
While all statins are associated with rare instances of rhabdomyolysis, a breakdown of muscle cells, Crestor had shown in studies before its approval that the potentially deadly disease had surfaced in seven people. Crestor’s potential muscle- and liver-damaging side effects become more worrisome and difficult to justify in patients who are essentially healthy.
What’s more, the study only lasted two years. That’s not long enough to adequately establish safety for the drug, especially if people are going to use it “preventatively”, which means they could be taking it for several years and even decades. Statins have caused cancer in every single animal study to date. Since cancer can take up to 25 years to develop after initial exposure to the carcinogen, we simply cannot know at this point that statins won’t also significantly increase the risk of cancer in adults.
Finally, before jumping on the statin bandwagon and recommending that we spend billions of dollars treating healthy people with Crestor, we should consider if there isn’t a less costly and risky way of preventing deaths due to inflammation and heart disease.
Wouldn’t you know it, there sure is!
For the last decade medical research has increasingly demonstrated that heart disease is caused not by high cholesterol levels, but by inflammation and oxidative damage. A full explanation of these mechanisms is beyond the scope of this post, but for more details you can read two previous articles: Cholesterol Doesn’t Cause Heart Disease and How To Increase Your Risk of Heart Disease.
So, if we want to prevent and even treat heart disease, we need to address the causes of inflammation and oxidative damage. Again, there’s not room to go into great detail on this here but in general the primary causes of inflammation and oxidative damage are 1) a diet high in polyunsaturated oil (PUFA) and refined flour and sugar, 2) lack of physical activity, 3) stress and 4) smoking.
We can thus prevent heart disease by avoiding PUFA and refined/processed food, getting adequate exercise, reducing stress and not smoking. These simple dietary and lifestyle changes are likely to produce even better results than a statin, for a fraction of the cost and without any side effects. In fact, the only side effects of this approach are improved physiological and psychological health! For more specific recommendations, read my article Preventing Heart Disease Without Drugs.
Taking a statin to “prevent” inflammation and heart disease is rather like bailing water with a pail to prevent a boat from sinking instead of simply plugging the leak. Unfortunately, our entire health care system is oriented around “bailing water with a pail”, which is to say treating the symptoms of disease, instead of “plugging the leak”, or addressing the causes of disease before it develops. The reason this is the case is because there’s a lot more money to be made from drugs, surgery and other costly interventions than there is from encouraging people to eat well, exercise and reduce stress.
Even if we ignore all of the issues I’ve pointed out above, the best thing we can say about this study is that a small group of unusual patients – those with low LDL-cholesterol AND high C-reactive protein – may slightly decrease their risk for all-cause mortality by taking a drug that costs them almost $1,300 per year and slightly increases their risk for developing diabetes.
That’s the best spin possible given the data from this study. Compare that to the mainstream media headlines, and you’ll have a clear understanding of how financial conflicts of interest are seriously damaging the integrity and value of clinical research.
At least the media wasn’t completely fooled. They did manage to at least include the perspective of sane doctors who questioned the desirability of millions of relatively healthy people taking drugs for the rest of their lives. According to the Wall Street Journal:
Moreover, despite large relative benefits, the actual number of patients helped was small. Those on the drug suffered 142 major cardiovascular events compared with 251 on placebo, a difference of 109. Dr. Hlatky said that raises questions about the cost-effectiveness of CRP screening and the value of putting millions of low-risk patients on medication for the rest of their lives.
From the New York Times:
Some consumer advocates and doctors raised concerns about the expense of putting relatively healthy patients on statins, which would cost the health system billions of dollars.
From Fox News:
About 120 people would have to take Crestor for two years to prevent a single heart attack, stroke or death, said Stanford University cardiologist Dr. Mark Hlatky. He wrote an editorial accompanying the study published online by the New England Journal of Medicine.
“Everybody likes the idea of prevention. We need to slow down and ask how many people are we going to be treating with drugs for the rest of their lives to prevent heart disease, versus a lot of other things we’re not doing” to improve health, Hlatky said.
If you know of someone who is considering a statin after reading about the JUPITER study, please do them a favor and send them a link to this article first. They should hear both sides of the story before making such a significant decision.“Everybody likes the idea of prevention. We need to slow down and ask how many people are we going to be treating with drugs for the rest of their lives to prevent heart disease, versus a lot of other things we’re not doing” to improve health, Hlatky said.
==============================================================
Read the full article here.














